
Research Article

Sequential immunoaffinity-LC/MS assay for quantitation of a therapeutic protein in monkey plasma
Linzhi Chen*, David Roos, Elsy Philip, Shirin Pagels
Boehringer Ingelheim Pharmaceuticals Inc., Ridgefield, CT, USA.
Vol.3, No. 5, Pages 127-138, doi: 10.17145/jab.17.016. (ISSN 2405-710X). Download PDF
Correspondence
Drug Metabolism and Pharmacokinetics US, Boehringer Ingelheim Pharmaceuticals Inc., 900 Ridgebury Road, Ridgefield, CT 06877, USA. Phone: +1 2037787870; Fax: +1 2037916003. Email: This email address is being protected from spambots. You need JavaScript enabled to view it.
Open Access and Copyright
©2017 Chen et al. This article is an open- access article distributed under the terms of the Creative Commons Attribution License (CC-BY) which permits any use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abstract
Immunocapture-LC/MS has recently been used for quantitating therapeutic proteins/peptides and biomarkers in various matrices. The advantages of LC/MS quantitation include high specificity and selectivity, wide dynamic range, and less susceptibility to interference from endogenous matrix components. We present a highly sensitive sequential immunoaffinity-LC/MS assay for quantitation of a biotherapeutic protein (39 kD) in monkey plasma. The first immunocapture utilized a biotinylated mouse anti-drug antibody to capture the drug in plasma. After tryptic digestion, a unique peptide from the drug was then captured by the second immunocapture using a mouse anti-peptide antibody for further sample purification. Samples analysis was performed on a microLC-triple quadrupole mass spectrometry system (MS/MS). Both immunocapture procedures were carried out in 96-well plates using a magnetic beads handler. The LLOQ of the assay is 50 pg/mL, which was approximately 100x more sensitive than a corresponding single immunocapture-LC/MS assay either using the anti-drug or anti-peptide antibody.
Keywords: anti-drug antibody, anti-peptide antibody, sequential immunoaffinity, immunocature-LC/MS, surrogate peptide.
Introduction
Quantitation of therapeutic proteins/peptides has been traditionally performed using ligand-binding assays (LBA) in support of drug discovery and development [1,2]. LBA relies on specific binding of target biotherapeutic analyte to capture /detection antibody to select and eventually detect the therapeutic in a complex matrix such as plasma. The most significant benefits of using LBA include high sample throughput and sensitivity. However, LBA is subject to interferences from endogenous matrix components that impact assay selectivity. In recent years, LC/MS has emerged as a promising platform for quantitation of biotherapeutics and protein biomarkers in biological matrices [3-6]. The vast majority of LC/MS-based protein quantifications are performed at peptide levels, mainly due to consideration of assay sensitivity [7-10]. A typical procedure for LC/MS-based quantification includes enzyme digestion and quantification of the target proteins based on selected surrogate peptides derived from the target [8,9]. Recently, immunocapture sample purification and enrichment prior to digestion and subsequent LC/MS analysis has been implemented [10-16]. The combination of immunocapture with LC/MS not only can address the selectivity issues affecting LBA, but also add significant benefits such as multiplexing (multiple analytes in one assay; same assay for multiple species/matrices), fast method development, wide dynamic range (up to 4 orders of magnitude), and good robustness and reproducibility.
For most immunocapture-LC/MS analysis of biotherapeutics, the immunocapture step is performed at the protein level [17]. It takes advantage of the unique immunoaffinity of the biotherapeutic and the capture agent, and thus provides unique selectivity. The capture agent, which is usually an antibody or target, is immobilized on magnetic beads or other solid support. During the immunocapture process, magnetic beads containing captured biotherapeutic are separated from matrix. After washing, the biotherapeutic is eluted from the beads by the addition of acid, followed by digestion and subsequent LC/MS analysis. As expected, the immunocapture sample cleanup produces a clean, enriched extract and greatly reduces matrix effect on the LC/MS analysis resulting in improved assay sensitivity [10, 13]. In an early example of such applications, a lower limit of quantitation (LLOQ) of low ng/mL was achieved for biotherapeutics such as Erbitux, a human:murine chimeric mAb used for the treatment of colorectal cancer [18].
An alternative approach involves immunocapturing a surrogate peptide of the protein biotherapeutic using an anti-peptide antibody after the sample is enzymatically digested. This approach was initially developed for quantitation of endogenous peptides and proteins, termed Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA) [19,20]. The peptide-level immunocapture can be carried out either offline [21-27] or online coupled to LC/MS [19,28−32], and has been mostly used for protein biomarker analysis.
Recently, sequential protein and tryptic peptide immunoaffinity LC/MS methodologies have been developed for highly specific and sensitive measurements of human β‑nerve growth factor (NGF) [33] as well as human and monkey interleukin-21 (IL-21) [34]. The workflow included offline enrichment of target protein using a capture antibody, followed by isolation using magnetic beads, trypsin digestion, online enrichment of tryptic peptides derived from target protein using anti-peptide antibodies, and quantification using nanoflow LC-MS/MS. An LLOQ as low as 7.03 pg/mL for NGF and 0.78 pg/mL for IL-21 was enabled using the sequential immunoaffinity approach. Robotic sample preparation and a robust chromatography configuration enabled this technology to be used for routine clinical analysis [34].
Here we describe a highly sensitive sequential immunoaffinity-LC/MS assay for quantitation of a protein biotherapeutic in monkey plasma. The first immunocapture is to capture and isolate the protein drug from monkey plasma, followed by tryptic digestion. A unique tryptic peptide derived from the drug is then captured in the second immunocapture process using an anti-peptide Ab for further sample purification. Both the protein and peptide immunocapture was performed offline in 96-well plates in order to increase throughput as well as reproducibility. Samples are injected onto a microflow-LC/MS for quantitation of the protein using the unique peptide as a surrogate.
Materials and Methods
Reagent and materials
Drug A is a proprietary experimental biotherapeutic protein of Boehringer Ingelheim Pharmaceuticals, Inc. (Ridgefield, CT, USA) and was produced in-house. It was recombinant consisting of 3 subunits and containing no human Fc, with a molecular weight of ~39 kDa. Surrogate peptides EGVSAIR and YDAVSLEGR, and their respective stable isotope labeled internal standards (IS), EGVSAI[13C6,15N4-R] and YDAVSLEG[13C6,15N4-R], were synthesized at Genscript (Piscataway, NJ, USA). Mouse monoclonal anti-drug antibody (mAb) 4E12, 5A5, 5A12, 2A8, 5A1, 4F1, 4B2, and 1D8 were supplied in-house. Mouse monoclonal anti-peptide YDAVSLEGR antibody 2E12, 4A3, 8B2, 9C2 and 9C10 were produced by Genscript. TPCK trypsin and EZ-Link Sulfo-NHS-LC biotinylation kits were obtained from Thermo Scientific (Rockford, IL, USA). Streptavidin magnetic beads (Magnesphere Paramagnetic Particles) were obtained from Promega (Madison, WI, USA). Cynomolgus monkey plasma was purchased from Bioreclamation (Westbury, NY, USA). All other lab chemicals, reagents and buffer solutions were obtained from Sigma Aldrich (St. Louis, MO, USA), Thermo Scientific or Invitrogen (Grand Island, NY, USA).
Generation of anti-peptide antibody
To boost immune response, the two surrogate peptides, EGVSAIR and YDAVSLEGR, were conjugated to keyhole limpet hemocyanin (KLH) using glutaraldehyde. Mice were immunized separately with 50 µg of the conjugated peptides, followed by three boost immunizations at two-week intervals. Peptide EGVSAIR did not show any immune response after the final boost and was therefore discontinued. Cell electrofusion was performed for peptide YDAVSLEGR at an average fusion efficiency of about one hybridoma per 5000 B cells which gave approximately 2 x 104 clones. Five positive clones were selected for subcloning. Based on ELISA screening with the target peptide, two stable subclonal cell lines of each primary clone were chosen for monoclone cryopreservation. Antibody production was carried out in roller bottles and purified by Protein A/G affinity column chromatography. The final products were certified by quality control, including purity test by SDS-PAGE, concentration determination by absorption at OD280 nm and antigen reactivity by ELISA.
Biotinylation
Biotinylation of the mouse monoclonal antibodies (mAb) was performed using an EZ-Link SulfoNHS-LC biotinylation kit following the vendor instructions. Typical biotin incorporation was determined to be approximately 2-7 biotins per mouse mAb molecule.
Preparation of standards and quality controls
A stock solution of drug A was received at a concentration of 10.28 mg/mL. Plasma spiking standards at 100 and 1 µg/mL were prepared from the stock solution using blank monkey plasma as a diluent. A series of monkey plasma calibration standards ranging from 0.05 to 100 ng/mL were prepared from the plasma spiking standards via serial dilution. Quality control (QC) samples at 0.15, 15 and 75 ng/mL were also prepared from the plasma spiking standards.
Single protein-level immunocapture
A 500 µL aliquot of monkey plasma sample, 75 µL of 0.1 mg/mL biotinylated mouse mAb against drug A and 425 µL of Tris-buffered saline with 0.1% Tween-20 (TBS-T) were added to a 96-deepwell polypropylene plate. The plate was incubated at room temperature for 2 hrs. A 40 µL aliquot of freshly prepared 5 mg/mL magnetic beads solution was added to each sample and the plate was gently mixed for 1 hr at room temperature. The beads were separated via magnet, washed three times with 300 µL of TBS-T and once with 300 µL of water, and then eluted with 100 µL of 25 mM HCl on a Kingfisher Flex magnetic bead handler. The eluent was immediately neutralized with 20 µL of 1 M Tris-HCl (pH 8.0). A 5 µL aliquot of 100 mM TCEP in 100 mM ammonium bicarbonate was added to each sample and the plate was incubated for 1 hr at 60°C. Next, 5 µL of 200 mM iodoacetamide in 100 mM ammonium bicarbonate was added to each sample and the plate was incubated for 30 minutes at room temperature in the dark. Five (5) µL of working internal standard (1,000 ng/ml in water), 5 µL of 100 mM CaCl2, and 5 µL of 6 µg/µL trypsin in 50 mM acetic acid were then added to each sample. The plate was incubated overnight at 37°C with gentle shaking. The digestion was quenched with 10 µL of 20% formic acid and the plate was centrifuged at 3800 rpm for 10 minutes before injection onto LC/MS for analysis.
Single peptide-level immunocapture
A 50 µL aliquot of monkey plasma and 50 µL of 16M urea were mixed in a 96-well plate for 30 minutes at room temperature. Fifty (50) µL of 100 mM TCEP in 100 mM ammonium bicarbonate was added to each sample and the plate was incubated at 60°C for 1 hr with gentle mixing. Next, 50 µL of 200 mM iodoacetamide in 100 mM ammonium bicarbonate was added to each sample and the plate was incubated for 30 minutes at room temperature in the dark. A 600 µL aliquot of trypsin working solution (0.2 mg/mL trypsin and 5 µM CaCl2 in 100 mM ammonium bicarbonate) was added to each sample for overnight digestion at 37°C. On the following day, 40 µL of 0.1 mg/mL biotinylated mouse anti-peptide antibody, 5 µL of working internal standard solution (1,000 ng/mL in water) and 160 µL of TBS-T were added to each sample. The plate was incubated at room temperature for 2 hrs. A 20 µL aliquot of freshly prepared 5 mg/mL magnetic beads was added to each sample and the plate was gently mixed for 1 hr at room temperature. After separating and washing the beads, the surrogate peptide was eluted from the beads and injected onto LC/MS for analysis.
Sequential immunoaffinity
The sequential immunoaffinity process consisted of protein–level immunocapture, trypsin digestion and peptide-level immunocapture in a sequential manner. The protein–level immunocapture procedure was same as the single protein-level immunocapture procedure described above. Following the overnight plasma digestion, 325 µL of TBS-T, 5 µL of working internal standard solution (1000 ng/mL in water) and 30 µL of 0.1 mg/mL biotinylated mouse anti-peptide antibody were added to each sample. The plate was then incubated at room temperature for 2 hrs. A 10 µL aliquot of freshly prepared 5 mg/mL magnetic beads was added to each sample and the plate was gently mixed for 1 hr at room temperature. After separating and washing the beads, the surrogate peptide was eluted from the beads and injected onto LC/MS for analysis.
LC/MS analysis
An Eksigent Ekspert MicroLC 200 coupled with an AB Sciex 6500 triple quadrupole mass spectrometer (AB Sciex, Framingham, MA, USA) was used. Chromatographic separation was performed using an Acquity Peptide BEH C18 column (1 mm x 50 mm, 1.7 µm, 300Å) operated at 60°C. Mobile phases consisted of (A) 0.1% formic acid and (B) 0.1% formic acid in acetonitrile running at a flow rate of 60 µL/min. The LC gradient was held at 5% B for 1 minute and then raised to 30% B over 2.75 minutes. The column was then washed with 95% B for 1.25 minutes and re-equilibrated for 1 minute before the next injection. The mass spectrometer was operated in positive electrospray ionization mode. Key instrument parameters were: +5500 V electrospray voltage, 50 nebulizer gas units, 30 axillary gas units, 375°C ion source temperature, 8 collision gas units, and unit resolution on both Q1 and Q3. Multiple-reaction-monitoring (MRM) with parent-to-product ion transitions 366.2 → 446.2 for EGVSAIR, 371.2 → 456.2 for EGVSAI[13C6,15N4-R], 505.1 → 561.3 for YDAVSLEGR, and 510.1 → 571.3 for YDAVSLEG[13C6,15N4-R] were used. For identifying the surrogate tryptic peptides of drug A, a 10 µg/mL aqueous solution was digested and analyzed by LC/MS using information-dependent acquisition (IDA).
Assay qualification
Assay qualification involved testing several key parameters including accuracy, precision, and specificity, and was intended to establish confidence in assay performance for potential further assay validation according to Good Laboratory Practice (GLP). The drug A plasma calibrant concentrations were 100, 80, 50, 10, 5, 1, 0.5, 0.1 and 0.05 ng/mL. The calibration curve, including a blank (containing no drug) and a double blank (no drug, no internal standard), was analyzed in duplicate. QC samples fortified with 0.15 (low), 15 (medium), and 75 (high) ng/mL drug in cynomolgus plasma were tested using 4 replicates at each level in 2 separate batches run on different days. EGVSAIR was used as the surrogate peptide for quantification of drug A with the single protein-level immunocapture procedure, while YDAVSLEGR was used for quantitation with the single peptide-level immunocapture procedure and the sequential immunoaffinity procedure.
Results and discussion
Drug A was an experimental therapeutic protein intended to be given to humans via intra-vitreal injection. A cynomolgus monkey pharmacokinetic (PK) study was conducted with an intra-vitreal dose of 0.25 mg drug A per eye. Vitreous humor, aqueous humor, retina tissue and plasma samples were collected for the determination of drug levels in these matrices. It was expected that the drug would stay primarily in the eyes and slowly distribute to blood circulation. As such, plasma drug levels would be very low and a sensitive bioanalytical PK assay with a lower limit of quantitation (LLOQ) of ≤ 100 pg/mL was needed in order to measure the plasma drug levels.
The goal of the present work was to develop an immunocapture-LC/MS assay with an LLOQ of ≤ 100 pg/mL to support the monkey PK ocular study. A stepwise approach was implemented for the assay development, in which three different immunocapture approaches, namely protein level only, peptide level only and sequential immunoaffinity (protein level and peptide level), were developed sequentially.
For a therapeutic protein larger than 10 kDa, direct quantitation of the whole protein by LC/MS may not be sensitive enough to support various drug discovery and development programs. Instead, quantitation is usually based on surrogate peptide(s) derived from the protein via digestion [7-9]. Most quantitation is performed on triple quadrupole (QQQ) or hybrid quadrupole time-of-flight (qTOF) mass spectrometers coupled with liquid chromatography using multi-reaction-monitoring (MRM) of surrogate peptide(s) [8]. As drug A had a molecular weight about 39 kDa, quantitation via surrogate peptide(s) was chosen for developing the sensitive immunocapture-LC/MS assay. The selection of drug A surrogate peptides involved several steps. First, in silico trypsin digestion of drug A was performed to generate a list of potential peptide candidates. Peptide size was limited to 6 to 20 amino acid residues which was found to be the right balance between assay specificity/selectivity and sensitivity [35-37]. In addition, the surrogate peptides should not contain methionine which can be prone to oxidation. The surrogate tryptic peptides must be unique to drug A and should not be produced from sample matrix, even though most matrix components would be removed by immunocapture purification. A database alignment of the amino acid sequences of drug A was performed using Blast (Basic Local Alignment Search Tool) [38]. Visual assessment of endogenous monkey plasma protein matches enabled the unique regions to be confirmed. Seven peptides were found to meet these criteria.
Trypsin digestion of 10 µg/mL drug A in phosphate buffer was performed to verify which of those peptides were indeed formed and detected by LC/MS. Among these 7 peptides predicted in silico, 6 were formed via the neat solution digestion. Peptides EGVSAIR and YDAVSLEGR were the most sensitive with MRM parent-to-product 366.220 (2+) → 446.235 and 505.131 (2+) → 561.300, respectively. These two peptides were taken into further method development including immunocapture efficiency assessment and LC/MS optimization as discussed later on.
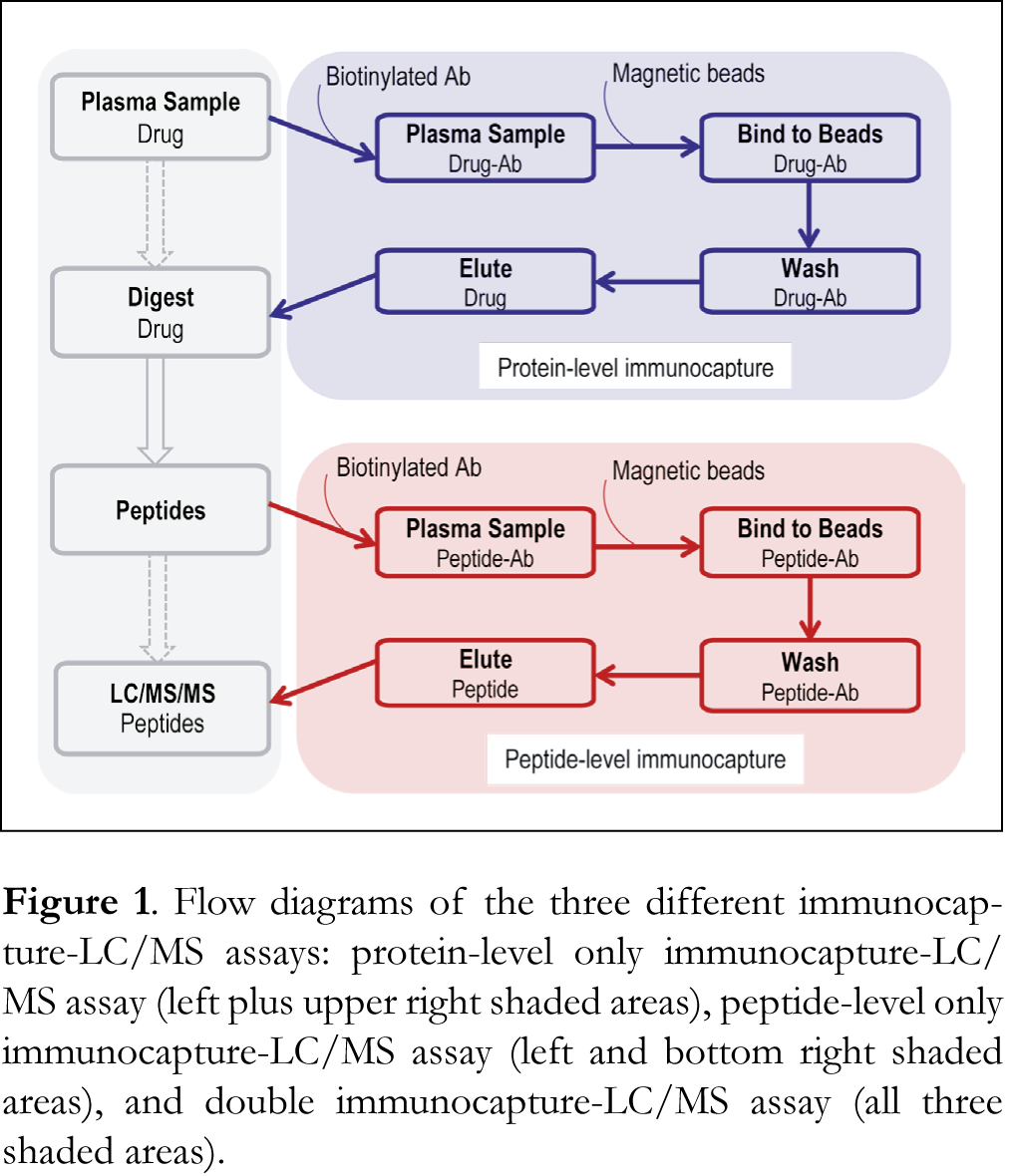

Micro-LC/MS with MRM detection
In the past two decades, LC/MS has become the most widely used bioanalytical technology in support of drug research and development. Electrospray ionization (ESI) [39] serves as a major interface between LC and MS. It is well understood that a greater sensitivity can be gained at reduced LC flow rate in ESI, particularly for macromolecules such as proteins [40,41]. Higher sensitivity and lower background has been largely realized by introducing miniaturization approaches into LC systems [42, 43]. Nano-flow LC/MS is popular for proteomic studies owing to its high sensitivity and drastically reduced sample volume consumption [44]. Micro-flow LC/MS can reach an optimal balance of sensitivity, throughput, and sample consumption, making it quite appealing and easy to adopt for bioanalytical laboratories [45]. It was reported that micro-LC/MS yielded a more than 14 x S/N improvement for analysis of methotrexate in human plasma when compared with the same method with the use of conventional HPLC–MS/MS [46]. Furthermore, the micro-LC/MS method was demonstrated to be as accurate and precise as the HPLC–MS/MS method.
For the LC/MS method development, we used an Eksigent Ekspert micro-LC 200 system, which operated at pressures up to 10000 psi and a flow rate of 5-200 μL/min. LC separation was performed on a reversed-phase Waters BEH C18 column (1 x 50 mm, 1.7 µm). A shallow gradient, 95-70% mobile phase A (0.1% formic acid) in 3.75 min at 60 µL/min, was employed. Sample injection volume was 10 µL, which despite being relatively large for micro-LC system still gave a very good peak. Adequate separation of the two peptides from background interference peaks was achieved under these LC conditions. The LC retention time was 2.88 min for EGVSAIR and 3.55 min for YDAVSLEGR. At the end of the gradient, the column was washed for 1 min at 95% B (0.1% formic acid in acetonitrile) to remove potential residues from previous samples. This washing step was found to be necessary in order to increase the assay reproducibility and robustness. The total run time was 6 minutes per sample, which presented a reasonable throughput.
To achieve maximal sensitivity, the Eskigent micro LC 200 system was coupled to a Sciex QTRAP 6500 triple quadrupole system. The LC/MS system was operated in positive ESI mode and optimized for the detection of peptides EGVSAIR and YDAVSLEGR. Parent-to-product ion pairs were optimized for the MRM detection. Both peptides were mostly doubly charged in ESI and cleaved to singly charged product ions upon collision activation. Product ions with m/z values higher than their respective parent ions were chosen so that any singly charged interferences would be eliminated from the detection. The most sensitive parent-to-product ion transition was 366.2 → 446.2 for EGVSAIR and 505.1 → 561.3 for YDAVSLEGR. Mid-resolution settings on both quadrupoles (Q1 and Q3) were used to minimize the interference peaks while not sacrificing instrument sensitivity.
For comparison, we also developed an LC/MS assay using a conventional Waters UPLC system coupled to the same QTRAP 6500 mass spectrometer. The UPLC method consisted of a gradient from 95-80% mobile phase A (0.1% formic acid in water) over 2 minutes and then 80-5% mobile phase A over another 0.5 minutes. The flow was held at 5% mobile phase A for 1.5 minutes followed by a 1 minute re-equilibration for a 5 minute total run time. The separation was performed on a reversed-phase Waters BEH C18 column (2.1 x 50 mm, 1.7 µm) at a flowrate of 0.3 mL/min. Upon injection of 10 pg each EGVSAIR and YDAVSLEGR on column, the micro-LC/MSMS system yielded approximately ~33x improvement in S/N for both peptides over the conventional UPLC-MS system.
Protein-level only immunocapture-LC/MS assay
LC/MS-based quantitation usually requires the use of IS to compensate for variability during sample preparation and LC/MS analysis. Ideally, a stable isotope-labeled version of analyte should be used as IS since it can track the analyte very closely in each step throughout the assay. For quantitation of small molecules, stable isotope-labeled IS are routinely used as they can be easily synthesized. Although stable isotope-labeled IS should be also used for protein analysis [47], such IS is usually not available. However, a rich body of literature has demonstrated that as long as validation tests demonstrate that the assay is reproducible and robust, there was no preference for the type of internal standard, such as stable isotope-labeled surrogate peptide, used for protein quantitation [48]. In this study, stable labeled peptides were used as IS.
The general workflow of the protein-level only immunocapture-LC/MS assay consisted of 6 steps: immunocapture, immobilization on beads, wash, elution, digestion and LC/MS detection (Figure 1). To achieve a maximal sensitivity, a large amount (500 µL) of monkey plasma was used. It should be noted that although this should not be an issue for human, monkey, dog or other large animals, the use of such a large amount of sample was impractical for smaller animals such as rodents. Stable isotope labeled IS, EGVSAI[13C6,15N4-R] and YDAVSLEG[13C6,15N4-R] , were added to the sample prior to overnight trypsin digestion. We compared digestion with and without prior denaturation in the presence of 8M urea and found that both gave similar LC/MS responses. As such, denaturation was not implemented in the assay. After quenching digestion with formic acid, the samples were injected onto the micro-LC/MS system for analysis. To achieve a high throughput, the experiments were carried out in 96-well plates and a KingFisher Flex system was used to automate all the magnetic bead handling steps.
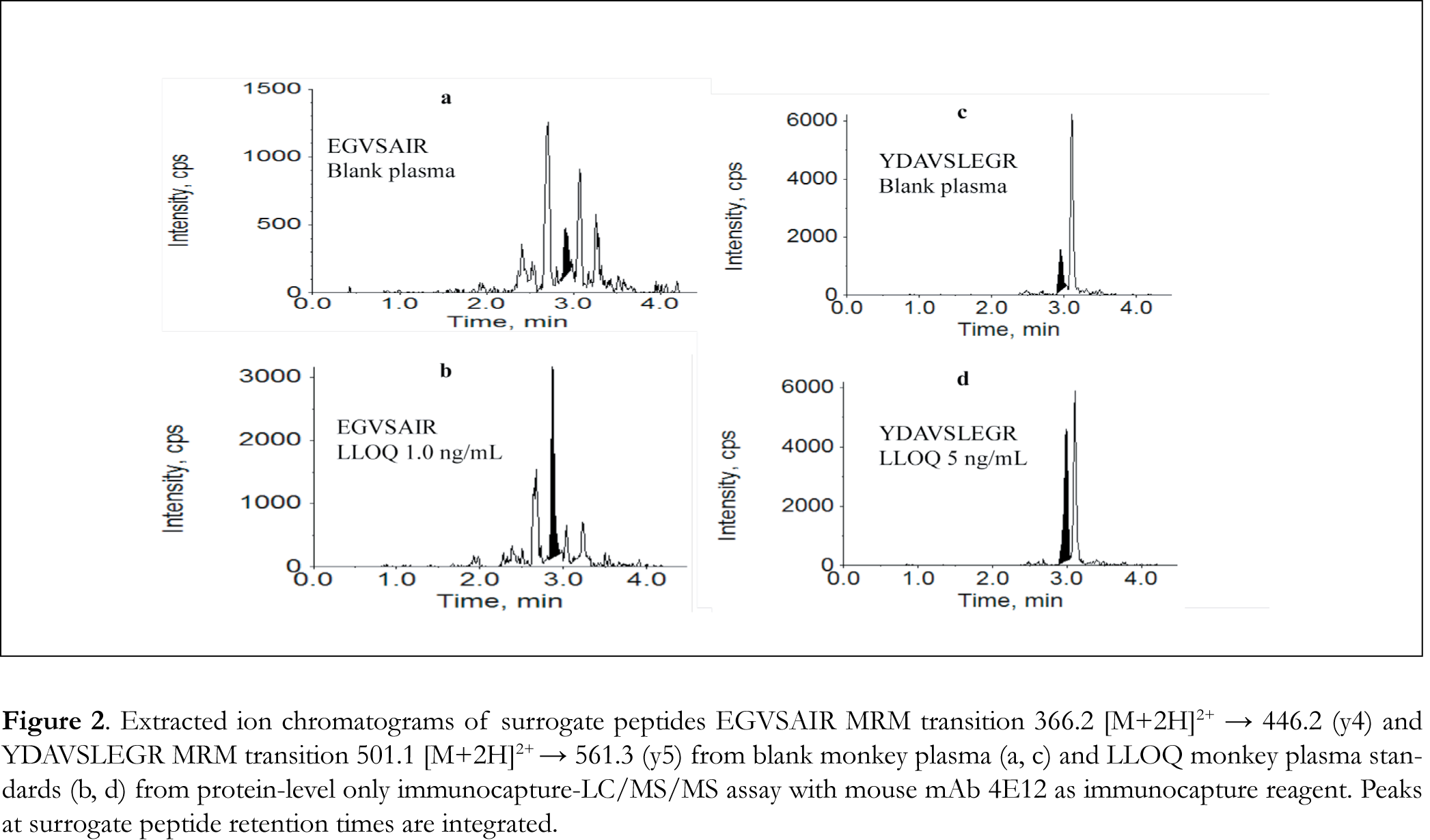
Eight mouse mAbs against drug A, 4E12, 5A5, 5A12, 2A8, 5A1, 4F1, 4B2, and 1D8, were tested for protein-level immunocapture. Five µL of 1 µg/µL mAb solution was added to 500 µL of 50 ng/mL neat drug A buffer solution and the sample was processed and assayed following the protein-level immunocapture-LC/MS procedures. The resultant LC/MS peak areas of the surrogate peptides EGVSAIR and YDAVSLEGR were used to evaluate immunocapture efficiency. All of the 8 mouse mAb were found to be able to capture drug A at different degrees. Capture antibody 4E12 gave the highest LC/MS response for both EGVSAIR (13900 peak area) and YDAVSLEGR (6290), representing the best immunocapture efficiency. Furthermore, the specificity/selectivity of the mAb was assessed using blank monkey plasma processed following the immunocapture procedures using the 8 different mAb as the immunocapture reagent. The LC/MS chromatograms showed small interference peaks at retention times of interest for both EGVSAIR and YDAVSLEGR with all of the 8 mAbs. The least interference was seen from 4E12 (Figure 2a,c) with peak heights of 511 and 1570 counts per seconds (cps) and peak area counts of 1070 and 7100 for EGVSAIR and YDAVSLEGR, respectively. Although the 2 peptides should not be produced from endogenous proteins in monkey plasma based on the BLAST search, any matrix residue on the magnetic beads might be carried over through digestion and contribute to the background responses. With the presence of the interferences, the attainable LLOQ was 1.0 ng/mL for EGVSAIR and 5.0 ng/mL for YDAVSLEGR using mAb 4E12 as the immunocapture reagent (Figure 2b,d), which were higher than the 100 pg/mL LLOQ required.
Peptide-level only immunocapture-LC/MS assay
Similar to the protein-level immunocapture, the peptide immunocapture approach relied on quality anti-peptide Ab. To produce anti-peptide Ab, ten mice were immunized with peptides EGVSAIR and another ten were immunized with YDAVSLEGR. The peptides were conjugated to keyhole limpet hemocyanin (KLH) to boost immune-response. While peptide YDAVSLEGR was highly immunogenic, peptide EGVSAIR did not elicit any immune-response after a primary immunization and subsequent 3 boost injections. Therefore, only peptide YDAVSLEGR was suitable for the peptide-level immunocapture approach. Among the 10 mice immunized with YDAVSLEGR, 5 produced Ab with reasonable affinities in an ELISA test. These 5 Abs (2E12, 4A3, 8B2, 9C2 and 9C10) were then biotinylated, spiked to 100 µl of a 500 ng/mL YDAVSLEGR buffer solution, and carried through the peptide-level immunocapture-LC/MS assay to assess their immunocapture efficiency. Compared to the starting amount of the peptide, 49.3% - 84.0% peptide were recovered through the immunocapture process with 9C10 being the highest while 4A3 being the lowest. Thus, 9C10 was chosen as the peptide-level capture reagent.
The peptide-level only immunocapture was performed after direct crude monkey plasma digestion (Figure 1). Plasma is a very complex matrix that contains several hundreds of thousands of proteins in a wide concentration range [49]. The total concentration of plasma proteins is approximately 60-80 mg/mL, with albumin and globulins being the most abundant. In consideration of such high amounts of endogenous proteins, only 50 µL plasma was used, which was also manageable in 96-well plate format. Using more plasma sample could yield a lower LLOQ but was not practical due to limitation of the plate well capacity. Furthermore, it was found that using a larger amount of plasma produced a large and irregular amount of precipitation which resulted in poor reproducibility. The IS (labeled YDAVSLEGR) was added prior to the peptide immunocapture step so that it tracked the immunocapture step in addition to the downstream sample preparation and LC/MS analysis.
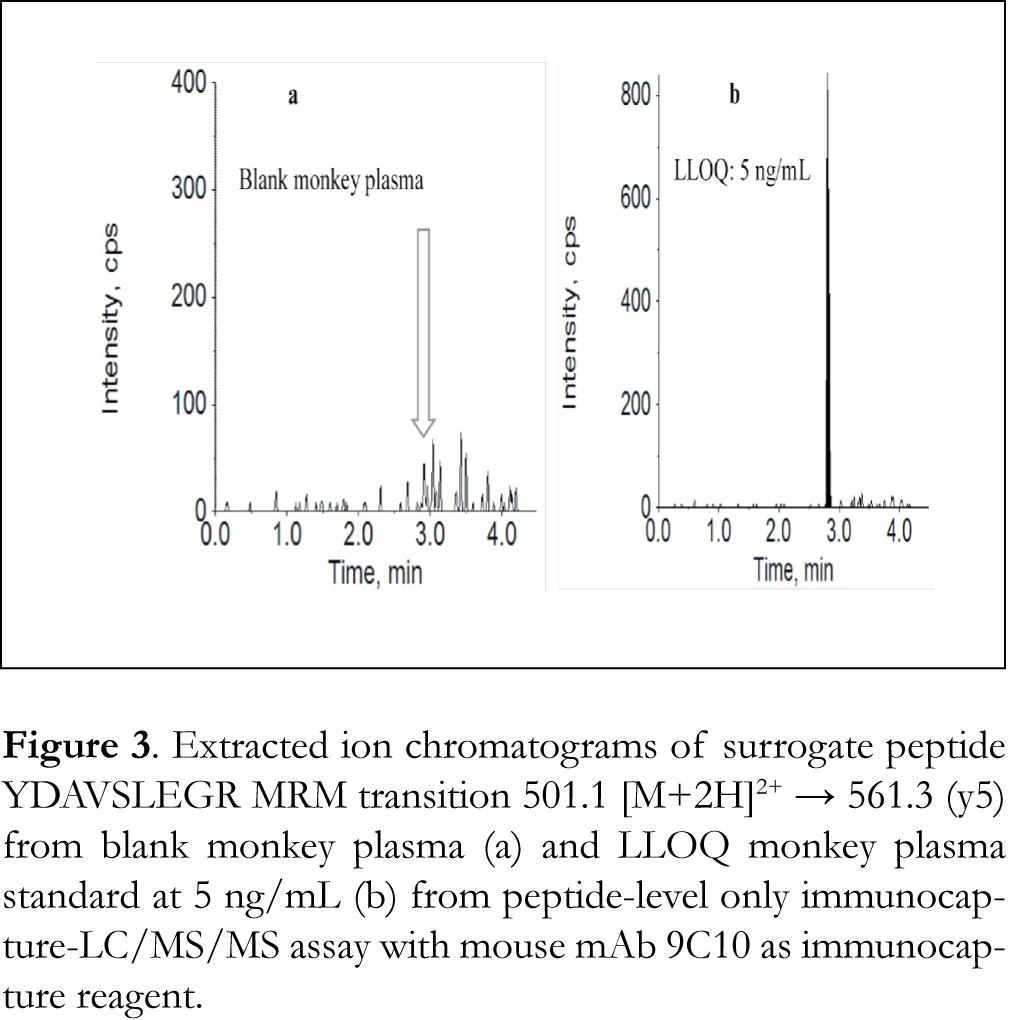
A typical LC/MS chromatogram of blank monkey plasma processed with the peptide-level immunocapture was shown in Figure 3a. A minor interference peak (50 cps) was observed at the retention time of peptide YDAVSLEGR. Compared to the protein-level immunocapture, the interference peak was much smaller. The assay dynamic range was 5 – 500 ng/mL with an LLOQ of 5 ng/mL and r2 = 0.9982. The LLOQ using peptide YDAVSLEGR as surrogate was the same as that from the protein-level immunocapture, even though only one-tenth of sample volume was used. Same as the protein-level only immunocapture, the peptide-level only immunocapture did not provide the required LLOQ of ≤ 100 pg/mL.
Sequential immunoaffinity-LC/MS assay
Since neither the protein-level only nor the peptide-level only immunocapture approach was able to provide the required assay sensitivity, our next attempt was to implement the two immunocapture processes in one assay. Figure 1 illustrates the sequential immunoaffinity workflow. Aliquots of 500 µL monkey plasma were used in the protein-level immunocapture. After capture, separation and enrichment from plasma at the protein-level using Ab 4E12 and streptavidin magnetic beads, drug A was digested overnight. The IS, YDAVSLEG[13C6,15N4-R], was added after the protein immunocapture and prior to digestion. The resulting peptide YDAVSLEGR along with the IS were captured using Ab 9C10, bound to magnetic beads and separated from matrix prior to LC/MS analysis.
The sequential immunoaffinity-LC/MS assay was optimized for magnetic bead type and amount as well as Ab amount at both the protein and peptide-level immunocapture. Two popular magnetic beads brands, Promega MagneSphere and Pierce Streptavidin, were tested. It was found that the Promega MagneSphere beads (1 µm dia) had the higher capacity and also resulted in less interference for both protein and peptide immunocapture. The optimal bead amount was 200 µg for the protein immunocapture and 50 µg for the peptide immunocapture. Likewise, the optimal Ab amount was 7.5 µg of Ab 4E12 for the protein immunocapture and 3 µg of Ab 9C10 for the peptide immunocapture.
Magnetic beads have large porous surfaces and are subject to nonspecific binding. A small amount of endogenous matrix components bound to the beads via nonspecific binding could interfere with the assay [50] if carried further onto digestion. To remove residual matrix, the beads were washed three times with 300 µL of TBS-T and once with 300 µL of water. After washing, the captured protein or peptide was eluted using 100 µL of 25 mM HCl. Under these acidic conditions, the antibody–antigen bond (dissociation constant Kd ~10-10 M) was cleaved and the captured protein or peptide was then released. On the other hand, the biotinylated capture antibodies 4E12 and 9C10 were bound to the beads via streptavidin–biotin interaction which has a higher affinity (Kd ~10-15 M) and should not be cleaved.
It was labor intensive to transfer magnetic beads from plate to plate manually using a pipette and a magnetic block. Both immunocapture processes were thus performed in 96-well plates using a KingFisher Flex magnetic bead handler. The KingFisher Flex system not only automated the labor-intensive bead handling processes, but also gave better precision and accuracy compared to manual operation.
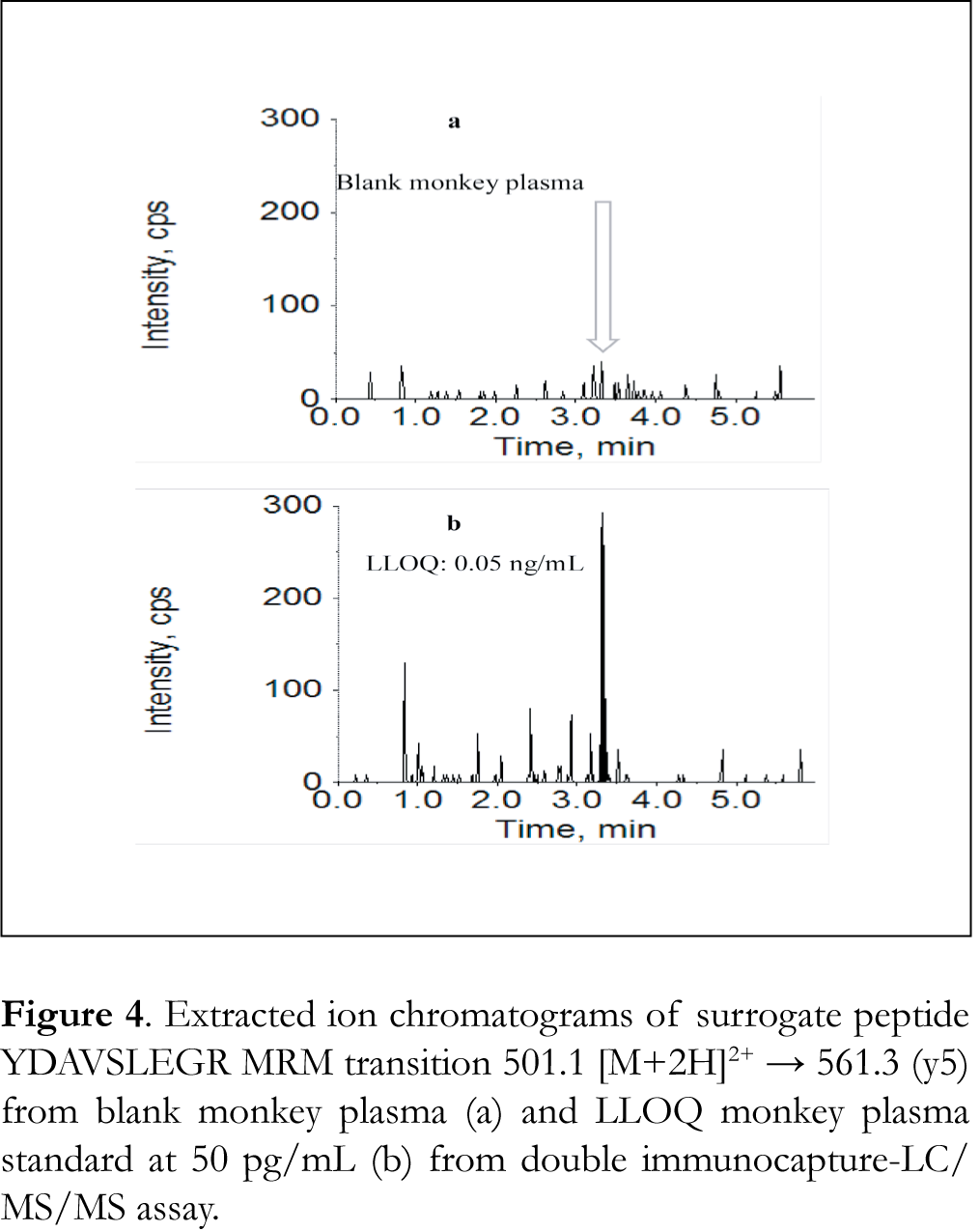
The assay produced an LLOQ of 50 pg/mL using 500 μL of plasma, which was 100x more sensitive than either the protein-level only or peptide-level only immunocapture with peptide YDAVSLEGR as the surrogate. Two major factors contributed to the assay sensitivity improvement. The starting plasma volume was 500 μL, same as for the protein-level only immunocapture but 10x more than for the peptide-level only immunocapture. The larger sample volume allowed for concentration of the final sample and an increase in sensitivity of the assay. The other factor was the reduced matrix interference. In the blank monkey plasma chromatogram (Figure 4a), nearly no interference peaks were observed at the retention time of peptide YDAVSLEGR. In comparison, considerable interference peaks were seen in the protein-level only immunocapture. Ten additional different lots of blank monkey plasma were tested and interferences in all lots were negligible. The achieved sensitivity is shown in Figure 4b, which displays the extracted ion chromatograms of the 50 pg/mL LLOQ drug A monkey plasma standard.
Assay linearity, accuracy and precision were assessed using plasma calibration standards and QC samples. Two batches were run on two different days to assess intra-batch (or intraday) and inter-batch (or inter-day) variability. Each curve was injected at the beginning and the end of each batch with 4 replicates of QC sets (low QC 0.15 ng/mL, medium QC 15 ng/mL and high QC 75 ng/mL QCs per set) in-between. A typical calibration curve obtained during assay qualification is shown in Figure 5. The assay dynamic linear range was examined by using peptide YDAVSLEGR/IS peak area ratios of nine calibration standards and applying a weighted (1/concentration2) least-squares linear regression. The assay linear range was determined to be 50 pg/mL – 100 ng/mL with r2 = 0.982. At the 50 pg/mL LLOQ plasma standard, peptide YDAVSLEGR peak height and area were 292 cps and 777, respectively. Mean back-calculated calibration standards were within ±15% of nominal values at all levels and CVs (coefficients of variation) were within 20% (data not shown). Summary statistics of intra- and inter-batch precision and accuracy of QC samples is presented in Table 1. Accuracies of the QCs (% relative errors) were within ±20%, and CVs were within 20%, which were within the criteria for assay acceptance. Drug A was previously determined using a different assay to be stable to up to at least 5 repeated freeze/thaw cycles, 24 hours on the benchtop at room temperature, and stable at least 1 year in -80 degree freezer in monkey plasma [data not shown]. Carryover, which was assessed by injecting a reagent blank immediately after the upper limit of quantitation (HLOQ) plasma standard at 100 ng/mL, was 88% when compared to the LLOQ. After three consecutive injections of the reagent blank, the carryover was reduced to 18%. Special care should be taken to limit the impact of carryover by adding extra wash injections following high concentration samples and also by assaying samples in order of lower to higher concentration when possible.
Conclusions
Although immunocapture at either protein level or peptide level effectively removes biological matrix such as plasma and significantly improves LC/MS assay sensitivity, trace amounts of matrix components still survive the purification process and limit sensitivity gains. For quantitation of the biotherapeutic protein drug A (~39 kDa), matrix interference was observed in both the protein-level and peptide-level single immunocapture assay, more prominently for the protein-level immunocapture assay. The achievable LLOQ was 1 ng/mL for the protein-level single immunocapture assay (using peptide EGVSAIR as quantitation surrogate), and 5 ng/mL for the peptide-level single immunocapture assay (using peptide YDAVSLEGR as quantitation surrogate). The matrix interference was almost completely removed by implementing both the protein-level and peptide-level immunocapture in a sequential immunoaffinity-LC/MS assay. Compared to the peptide-level immunocapture, sequential immunoaffinity enabled the use of a much larger amount of plasma (500 µL) which resulted in 100x higher assay sensitivity. The bead-based sample preparation allows the antibody capture step to be done in parallel in 96-well plates for multiple samples using a KingFisher Flex automated magnetic beads handler, resulting in a relatively high throughput. The sequential immunoaffinity assay achieved an LLOQ of 50 pg/mL and a wide dynamic range (50 pg/mL – 100 ng/mL). The assay sensitivity met the LLOQ requirement (≤ 100 pg/mL) in support of monkey ocular studies in which drug A plasma level was expected to be very low. Albeit the success of this sequential immunoaffinity approach, generation of anti-peptide Ab could be a limiting factor. Some small peptides may not be immunogenic and do not elicit immune-response in mice or other animals. In this study, only one of the two most sensitive surrogate peptides was immunogenic and able to produce Ab in mice after 4 injections with drug A.
Acknowledgment
The authors would like to thank Drs. Jeff Duggan, Hongbin Yu, Christine Grimaldi and Cheikh Kane for their help during the study.
References
1. DeSilva B, Smith W, Weiner R et al. Recommendations for the bioanalytical method validation of ligand-binding assays to support pharmacokinetic assessments of macromolecules. Pharm. Res. 20(11), 1885–1900 (2003). [CrossRef]
2. Myler HA, Given A, Kolz H, Mora JR, Hristopoulos G. Biotherapeutic bioanalysis: a multi-indication case study review. Bioanalysis 3, 623–643 (2011). [CrossRef]
3. Kay RG, Gregory B, Grace PB, Pleasance S. The application of ultra-performance liquid chromatography/tandem mass spectrometry to the detection and quantitation of apolipoproteins in human serum. Rapid Commun Mass Spectrom 21(16), 2585–2593 (2007). [CrossRef]
4. Heudi O, Barteau S, Zimmer D et al. Towards absolute quantification of therapeutic monoclonal antibody in serum by LC-MS/MS using isotope-labeled antibody standard and protein cleavage isotope dilution mass spectrometry. Anal Chem 80, 4200-4207 (2008). [CrossRef]
5. Pan S, Aebersold R, Chen R et al. Mass spectrometry based targeted protein quantification: methods and applications. J of Proteome Res 8, 787-797 (2009). [CrossRef]
6. An B, Zhang M, Qu J. Towards sensitive and accu rate analysis of antibody biotherapeutics by LC/MS. Drug Metab Dispos 42(11), 1858-1866 (2014). [CrossRef]
7. Blackburn M. Advances in the quantitation of therapeutic insulin analogues by LC-MS/MS. Bioanalysis 5, 933-946 (2013). [CrossRef]
8. Lange V, Picotti P, Domon B, Aebersold R. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol Syst Biol 4, 222 (2008). [CrossRef]
9. Liebler DC, Zimmerman LJ. Targeted quantitation of proteins by mass spectrometry. Biochemistry 52, 3797-3806 (2013). [CrossRef]
10. Xu Y, Mehl JT, Bakhtiar R, Woolf EJ. Immunoaffinity purification using anti-PEG antibody followed by two dimensional liquid chromatography/tandem mass spectrometry for the quantification of a PEGylated therapeutic peptide in human plasma. Anal Chem 82, 6877–6886 (2010). [CrossRef]
11. Cingöz A, Hugon-Chapuis F, Pichon V. Total online analysis of a target protein from plasma by immunoextraction, digestion and liquid chromatography mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 878, 213–221 (2010). [CrossRef]
12. Chenau J, Fenaille F, Ezan E et al. Sensitive detection of Bacillus anthracis spores by immunocapture and liquid chromatography-tandem mass spectrometry. Anal Chem 83, 8675–8686 (2011). [CrossRef]
13. Wang W, Walker ND, Zhu LJ et al. Quantification of circulating D-dimer by peptide immunoaffinity enrichment and tandem mass spectrometry. Anal Chem 84, 6891–6898 (2012). [CrossRef]
14. Wang Y, Heilig JS. Differentiation and quantification of endogenous and recombinant-methionyl human leptin in clinical plasma samples by immunocapture/mass spectrometry. J Pharm Biomed Anal 70, 440–446 (2012). [CrossRef]
15. Torsetnes SB, LØvbak SG, Claus C et al. Immunocapture and LC–MS/MS for selective quantification and differentiation of the isozymes of the biomarker neuron-specific enolase in serum. J Chromatogr B Analyt Technol Biomed Life Sci 929, 125–132 (2013). [CrossRef]
16. Bronsema KJ, Bischoff R, Pijnappel WW, van der Ploeg AT, van de Merbel NC. Absolute quantification of the total and antidrug antibody-bound concentrations of recombinant human α-glucosidase in human plasma using protein G extraction and LC–MS/MS. Anal Chem 87, 4394–4401 (2015). [CrossRef]
17. Fung EN, Bryan P, Kozhich A. Techniques for quantitative LC–MS/MS analysis of protein therapeutics: advances in enzyme digestion and immunocapture. Bioanalysis 8(8), 847–856 (2016). [CrossRef]
18. Dubois M, Fenaille F, Clement G, Lechmann M, Tabet JC, Ezan E, and Becher F. Immunopurification and mass Spectrometric quantification of the active form of a chimeric therapeutic antibody in human serum. Anal Chem 80, 1737-1745 (2008). [CrossRef]
19. Anderson, NL, Anderson, NG, Haines, LR, Hardie, DB, Olafson RW, and Pearson TW. Mass spectrometric quantitation of peptides and proteins using stable isotope standards and capture by anti-peptide antibodies (SISCAPA). J Proteome Res 3, 235-244 (2004). [CrossRef]
20. Whiteaker JR, Zhao L, Zhang HY, Feng LC, Piening BD, Anderson L and Paulovich AG. Antibody-based enrichment of peptides on magnetic beads for mass-spectrometry-based quantification of serum biomarkers. Anal Biochem 362, 44–54 (2007). [CrossRef]
21. Kuhn E, Addona T, Keshishian H, Burgess M, Mani DR, Lee RT, Sabatine MS, Gerszten RE, Carr SA. Developing multiplexed assays for troponin I and interleukin-33 in plasma by peptide immunoaffinity enrichment and targeted mass spectrometry. Clin Chem 55, 1108–17 (2009). [CrossRef]
22. Ahn YH, Lee JY, Lee JY, Kim Y-S, Ko JH, Yoo JS. Quantitative analysis of an aberrant glycoform of TIMP1 from colon cancer serum by L-PHAenrichment and SISCAPA with MRM mass spectrometry. J Prot Res 8:4216 –24 (2009). [CrossRef]
23. Hoofnagle AN, Becker JO, Wener MH, Heinecke JW. Quantification of thyroglobulin, a lowabundance serum protein, by immunoaffinity peptide enrichment and tandem mass spectrometry. Clin Chem 54, 1796–804 (2008). [CrossRef]
24. Schoenherr RM, Zhao L, Whiteaker JR, Feng LC, Li L, Liu L, et al. Automated screening of monoclonal antibodies for SISCAPA assays using a magnetic bead processor and liquid chromatography-selected reaction monitoring mass spectrometry. J Immun Meth 353, 49 – 61 (2010). [CrossRef]
25. Whiteaker JR, Zhao L, Anderson L, Paulovich AG. An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol Cell Proteome 9, 184–96 (2010). [CrossRef]
26. Whiteaker JR, Zhao L, Zhang HY, Feng LC, Piening BD, Anderson L, Paulovich AG. Antibody based enrichment of peptides for massspectrometry-based quantification on magnetic beads of serum biomarkers. Anal Biochem. 362, 44 –54 (2007). [CrossRef]
27. Whiteaker, JR, Zhao L, Frisch C, Ylera F, Harth S, Knappik A, Paulovich SG. High-affinity recombinant antibody fragments (Fabs) can be applied in peptide enrichment immuno-MRM assays. J Proteome Res 13, 2187–2196 (2014). [CrossRef]
28. Neubert H, Gale J, Muirhead D. Online high-flow peptide immunoaffinity enrichment and nanoflow LC-MS/MS: assay development for total salivary pepsin/pepsinogen. Clin Chem 56(9), 1413–1423 (2010). [CrossRef]
29. Anderson NL, Jackson A, Smith D, Hardie D, Borchers C, Pearson TW. SISCAPA peptide enrichment on magnetic beads using an in-line bead trap device. Mol Cell Proteome 8, 995-1005 (2009). [CrossRef]
30. Berna M, Schmalz C, Duffin K, Mitchell P, Chambers M, Ackermann B. Online Immunoaffinity liquid chromatography/tandem mass spectrometry determination of a type II collagen peptide biomarker in rat urine: investigation of the impact of collision-induced dissociation fluctuation on peptide quantitation. Anal Biochem 356, 235–43 (2006). [CrossRef]
31. Nemirovskiy OV, Dufield DR, Sunyer T, Aggarwal P, Welsch DJ, Mathews WR. Discovery and development of a type II collagen neoepitope (TIINE) biomarker for matrix metalloproteinase activity: from in vitro to in vivo. Anal Biochem 361, 93–101 (2007). [CrossRef]
32. Radabaugh MR, Nemirovskiy OV, Misko TP, Aggarwal,P, Mathews WR. Immunoaffinity liquid chromatography-tandem mass spectrometry detection of nitrotyrosine in biological fluids: development of a clinically translatable biomarker. Anal Biochem 380, 68 –76 (2008). [CrossRef]
33. Neubert H, Muirhead D, Kabir M, Grace C, Cleton A, Arends R. Sequential protein and peptide immunoaffinity capture for mass spectrometry-based quantification of total human β-nerve growth factor. Anal Chem 85, 1719–1726 (2012). [CrossRef]
34. Palandra J, Finelli A, Zhu M, Masferrer J, Neubert H. Highly specific and sensitive measurements of human and monkey interleukin 21 using sequential protein and tryptic peptide immunoaffinity LC–MS/MS. Anal Chem 85, 5522–5529 (2013). [CrossRef]
35. Wu ST, Ouyang Z, Olah TV, Jemal M. A strategy for liquid chromatography/tandem mass spectrometry based quantitation of pegylated protein drugs in plasma using plasma protein precipitation with water-miscible organic solvents and subsequent trypsin digestion to generate surrogate peptides for detection. Rapid Commun. Mass Spectrom 25 (2), 281–290 (2011). [CrossRef]
36. Chen LZ, Roos D, Philip E. Development of an immunocapture-LC/MS assay for simultaneous ADA isotyping and semi-quantitation. J Immunol Res 2016, 1-4 (2016). [CrossRef]
37. Roos D, Chen LZ, Vesapogu R, Kane C, Duggan J, Norris S. Detection of cynomolgus monkey anti-protein XYZ antibody using immunocapture-LC/MS. J Appl Bioanal 2(4), 117-128 (2016). [CrossRef]
38. Basic Local Alignment Search Tool. http://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins. Last access on June 3rd, 2017.
39. Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science 246, 64−71 (1989). [CrossRef]
40. Emmett MR, Caprioli R M. Micro-electrospray mass spectrometry: Ultra-high-sensitivity analysis of peptides and proteins. J Am Soc Mass Spectrom 5, 605−613 (1994). [CrossRef]
41. Wilm, MS, Mann, M. Electrospray and Taylor-cone theory, Dole’s beam of macromolecules at last? Int. J. Mass Spectrom. Ion Proc 136, 167−180 (1994). [CrossRef]
42. Catherman AD, Skinner OS, Kelleher N. Top Down proteomics: facts and perspectives. Biochem Biophys Res Commun 445, 683−693 (2014). [CrossRef]
43. Gikaa HG, Theodoridisb GA, Plumbc RS, Wilson ID. Current practice of liquid chromatography–mass spectrometry in metabolomics and metabonomics. J Pharm Biomed Anal 87, 12−25 (2014). [CrossRef]
44. Kennedy RT, Jorgenson JW. Preparation and evaluation of packed capillary liquid hromatography columns with inner diameters from 20 to 50 micrometers. Anal Chem 61, 1128–1135 (1989). [CrossRef]
45. Arnold DW, Needham SR. Micro-LC–MS/MS: the future of bioanalysis. Bioanalysis 5(11), 1329-1331 (2013). [CrossRef]
46. Christianson CC, Johnson CJ, Needham SR. The advantages of microflow LC–MS/MS compared with conventional HPLC–MS/MS for the analysis of methotrexate from human plasma. Bioanalysis 5(11), 1387-1396 (2013). [CrossRef]
47. Bronsema KJ, Bischoff R, Van De Merbel NC. Internal standards in the quantitative determination of protein biopharmaceuticals using liquid chromatography coupled to mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 893-894, 1–14 (2012). [CrossRef]
48. Song A, Lee A, Garofolo F, Kaur S, Duggan J, et al. White Paper on recent issues in bioanalysis: focus on biomarker assay validation (BAV): (Part 2 – Hybrid LBA/LCMS and input from regulatory agencies). Bioanalysis 8(23), 2457–2474 (2016). [CrossRef]
49. Omenn GS, Menon R, and Adamski M. The human plasma and serum proteome. Proteomics 3, 195–224 (2007). [CrossRef]
50. Trufelli H, Palma P, Famiglini G, Cappiello A. An overview of matrix effects in liquid chromatography mass spectrometry. Mass Spectrom Rev 30, 491-509 (2011). [CrossRef]
All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License.
![]()
 Open Access Article
Open Access Article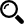 Peer-reviewed Article
Peer-reviewed Article Creative Commons Attribution 4.0 License
Creative Commons Attribution 4.0 License