
Research Article

Urine analysis of buprenorphine/norbuprenorphine/naloxone in drugs and driving cases
Albert A. Elian1, Jeffery Hackett2,*
1Massachusetts State Police Crime Laboratory, 14 Acton Street, Maynard, Massachusetts, MA 01754, USA.
2Forensic Laboratory Division, Office of Chief Medical Examiner, 850 Bryant Street, San Francisco, CA 94103, USA.
Vol.1, No. 3, Pages 80-88, doi: 10.17145/jab.15.014. (ISSN 2405-710X). Download PDF
Correspondence
*Correspondence: Forensic Laboratory Division, Office of Chief Medical Examiner, 850 Bryant Street, San Francisco, CA 94103, USA.
Phone: +1 415 553 9009; Fax: +1 415 553 9815. E-mail: This email address is being protected from spambots. You need JavaScript enabled to view it.
Open Access and Copyright
©2015 Elian AA and Hackett J. This article is an open access article distributed under the terms of the Creative Commons Attribution License (CC-BY) which permits any use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abstract
Buprenorphine is now being detected as a drug of abuse in drugs/ driving cases alongside side naloxone. Being a semi synthetic opiate, it has the potential to impair motorists in a similar fashion to morphine. Many laboratories have observed that buprenorphine does not produce a positive response with typical opiate immunoassays or routine basic GC-MS drug screens.
In this study, samples of urine were enzymatically hydrolyzed after which they were extracted on mixed mode solid phase (SPE) columns, after which the extracted samples were analyzed by LC-MS/MS analysis in positive multiple reaction monitoring mode.
The LOD/LOQ were determined to be 0.5 and 1.0 ng/ mL, respectively for the analytes; Linearity(10 to 1000 ng/ mL) and (r2>0.999). The recovery of the analytes was found to be greater than 90%. Interday/ Intraday analysis was found to < 8% and < 10 %, respectively. Matrix effects were determined to be < 6%.
Keywords: buprenorphine, norbuprenorphine, naloxone, LC-MS/MS.
Introduction
Buprenorphine (2S)-2-[(5R,6R,7R,14S)-9α-Cyclopropylmethyl-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxymorphinan-7-yl]-3,3-dimethylbutan-2-ol) is a semi-synthetic opioid, It possesses partial agonist properties acting at µ-opioid receptor site, it also possesses antagonist characteristics acting mainly on the κ-opioid receptor site [1]. Its structure is derived from thebaine, and it has a structural similarity with morphine ((5α,6α)-7,8-didehydro-4,5-epoxy-17-methylmorphinan-3,6-diol),but has been reported to be up to 50 times more potent, with a higher affinity for µ-opioid receptors compared with other opioids, including heroin (diacetyl morphine) [2]. Historically, buprenorphine was first synthesized 1966 by John Lewis working at Reckitt and Colman (later Reckitts). Lewis had previously been a doctoral student of Sir Robert Robinson, who had elucidated the active structure of morphine in 1925 [3]. Buprenorphine (BUP) is metabolized in human beings through to the active metabolite norbuprenorphine (NBUP) via the process of N-dealkylation, performed primarily by cytochrome P450 (CYP 450) 3A4 and CYP2D6 groups of enzymes [4]. Minor metabolites of BUP and NBUP i.e hydroxybuprenorphine and hydroxynorbuprenorphine have also been reported [5], these are believed to occur after oxidation of the tertiary butyl group on both BUP and NBUP but do not make a significant contribution to the urinary profile. Peak plasma concentration times of BUP have reported to range from from 0.66 hours to 3.5 hours, its half-life (t0.5) has been reported to be as long as 44 hours [6].
NBUP the primary metabolite of buprenorphine is by nature is a weak opiate agonist and has been reported as having a potency of one quarter of buprenorphine. It has also been reported that it possesses greater respiratory depressant effects than the parent and this phenomenon may controlled not by brain based opioid receptors but those located in the lung [7]. The half-life for NBUP has been reported as being longer than that of the parent by a factor of 1.5 [8].
BUP is now being detected as a drug of abuse in drugs and driving cases [9]. This drug is also found along in medications with in combination with naloxone (Suboxone®). Naloxone (NALOX) is described as a non-selective opiate antagonist at µ, κ, and δ opioid receptors. NALOX is added to BUP formulations to prevent the misuse by individuals wishing to administer the drug via an intravenous route [10]. NALOX is also an antagonist, with a lower affinity for the κ-and δ-opioid receptors. Naloxone is used to reverse the effects of opiate overdose, such as respiratory depression, sedation and hypotension, without a risk of developing tolerance to this particular drug. The final step in the metabolic process for the three compounds is the formation of their individual glucuronides [10]. Naloxone is not within the testing regime of many forensic toxicology laboratories in USA, any sample requiring testing for an analyte outside the laboratory’s testing manual would be referred to a reference testing facility under contract with the laboratory.
Neither BUP nor NBUP produce a positive response with typical opiate immunoassays, although many commercial ELISA manufacturers are now supplying BUP specific kits [11,12]. The parent and metabolite may not be detected by in basic drug screens after by using gas chromatography-mass spectrometry (GC-MS) without prior hydrolysis of the sample and post extraction derivatization for GC-MS in selected ion monitoring (SIM) mode, thus may not be detected in these routine drug screens. This is because the glucuronides are not as amenable as the parent compounds for analysis due to their difficulty in derivatizing for GC-MS analysis. This may answer the question of why BUP is being diverted into the illicit market as opiates are widely tested in forensic toxicology facilities but not BUP/NBUP.
In low concentrations, BUP/NBUP act like similar opiates/opioids in providing pain relieving or analgesic effects in subjects being administered the drugs, in high concentrations they also present similar adverse effects i.e. severe depression of the respiratory system, sedation and hypotension [13]. Thus the addition of NALOX is a preventative measure against overmedication.Previous methods published for the analysis of BUP/NBUP have employed the use of GC-MS [14-22], in these methods the compounds were derivatized after after being extracted from the urine samples using either liquid-liquid extraction [16] or solid phase extraction [15]. Typically, a silyl reagent such as BSTFA has been employed although an acyl derivative has been reported [22] as giving a better mass spectrometric performance. With the introduction of affordable liquid chromatography-tandem mass spectrometry (LC-MS/MS) units, reports have been published using this methodology [23-26]. Publications investigating the extraction and analysis of BUP, NBUP,and NALOX are not as populous as those involving buprenorphine (and its metabolite) and naloxone as singly entities. The nature of these studies tends to be of a clinical nature and not forensic toxicological one [27-33]. In this study, the authors have taken a forensic approach to apply the method to medico-judicial cases.
In this study urine samples taken from drugs and driving cases that previously tested positive for buprenorphine using LC-MS/MS were further analyzed for both the buprenorphine metabolite (norbuprenorphine) and naloxone to test the efficiency of the procedure. Urine is taken from suspects in some states such as Massachusetts by enforcement agencies rather than blood. The validation procedure follows guidelines set out in the current quality manual at Massachusetts State Police Crime Laboratory (MSPCL).
Experimental.
Reagents and Equipment
Buprenorphine, norbuprenophine, buprenorphine-d3, norbuprenorphine-d4, and naloxone (1.0 mg/mL, 1.0 mg/mL, 0.1 mg/mL, 0.1 mg/mL, 1.0 mg/mL, respectively) were obtained from Lipomed, Cambridge MA). Acetonitrile, ammonium hydroxide, acetic acid (glacial), methylene chloride and methanol were obtained from Fisher Scientific (Pittsburgh, PA). Phosphate buffer (0.1 M, pH 6) and pH 5 acetate buffer (0.1M) were purchased ready prepared from Fisher Scientific. Formic acid was obtained from Acros Chemicals (via Fisher Scientific). DI (deionized) water was generated in house. All chemicals were of ACS grade. Solid phase extraction (SPE) columns (Clean Screen®DAU (6mL, 200 mg)), Selectrazyme β-glucuronidase (derived from Red Abalone) were obtained from UCT, Inc., (Bristol PA). Negative urine was obtained from volunteers and laboratory tested to be drug free.
Formic acid was made up as a 0.1% (v/v) solution by the addition of 1 mL of the acid to 900 mL of DI water and diluting to 1 L. Acetonitrile containing 0.1% (v/v) was made up by adding 1 mL of formic acid to 900 mL of acetonitrile and diluting to 1 L with the same solvent.
Analysis was performed using an API 3200 Q-Trap instrument supplied by Applied Biosystems (Foster City, CA). The chromatographic system consisted of a Shimadzu CBM 20 A controller, two Shimadzu LC 20 AD pumps including degasser, a Shimadzu SIL 20 AC autosampler, and a Shimadzu CTO AC autosampler compartment (set at 10˚C), the instrument was fitted with a Selectra DA column (50 x 2.1 mm (5 µm)) from UCT Inc), and was attached to a Selectra DA guard column (10 x 2mm) which was obtained from the same supplier.
Liquid Chromatography-Mass Spectrometry
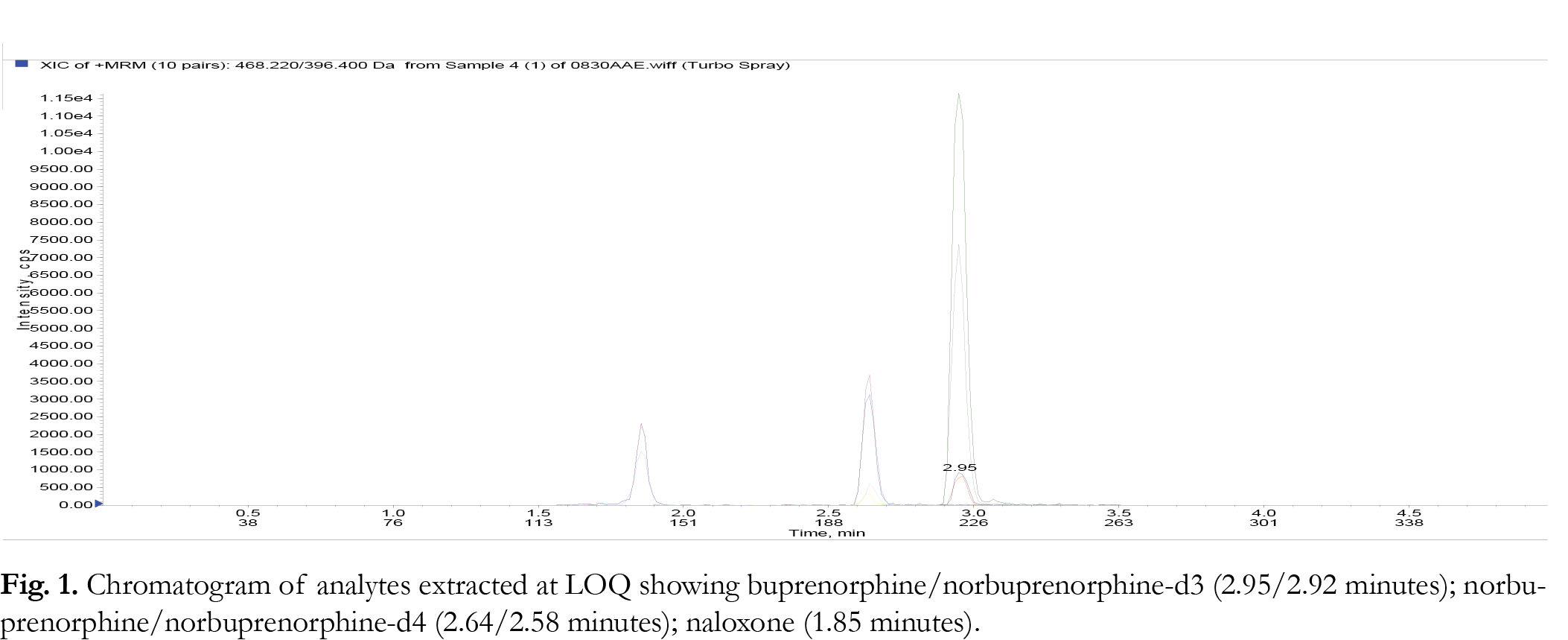
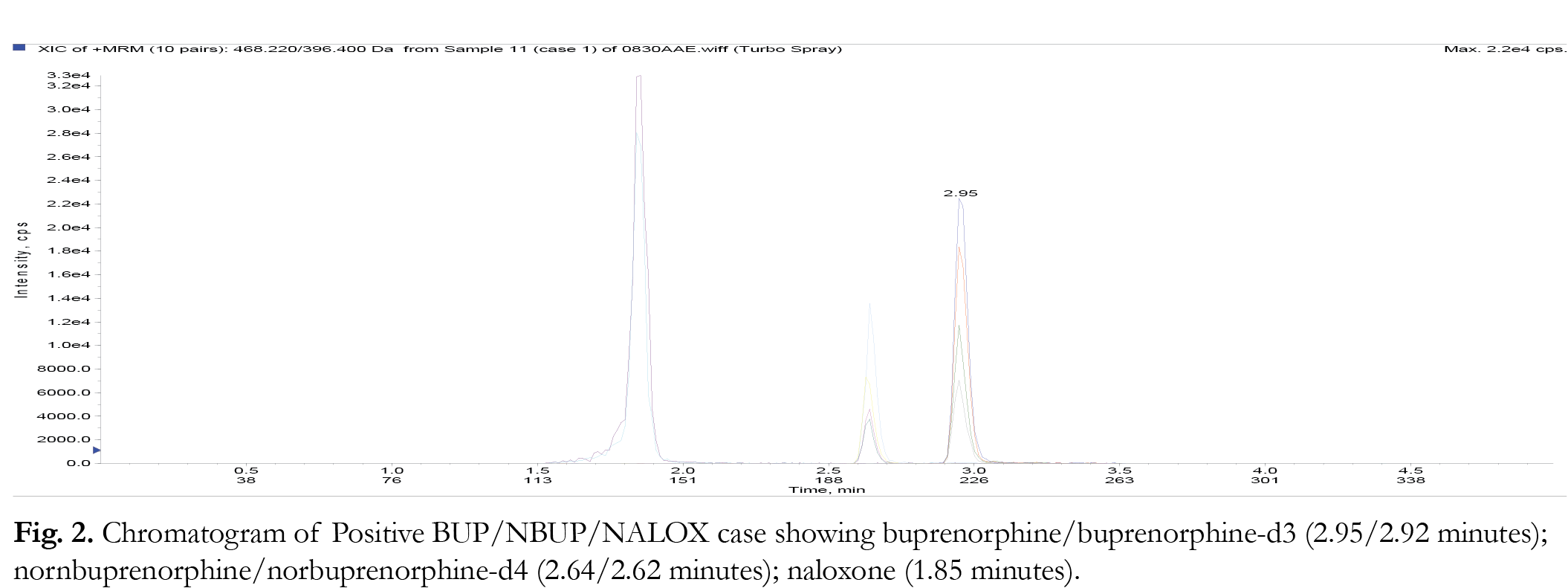
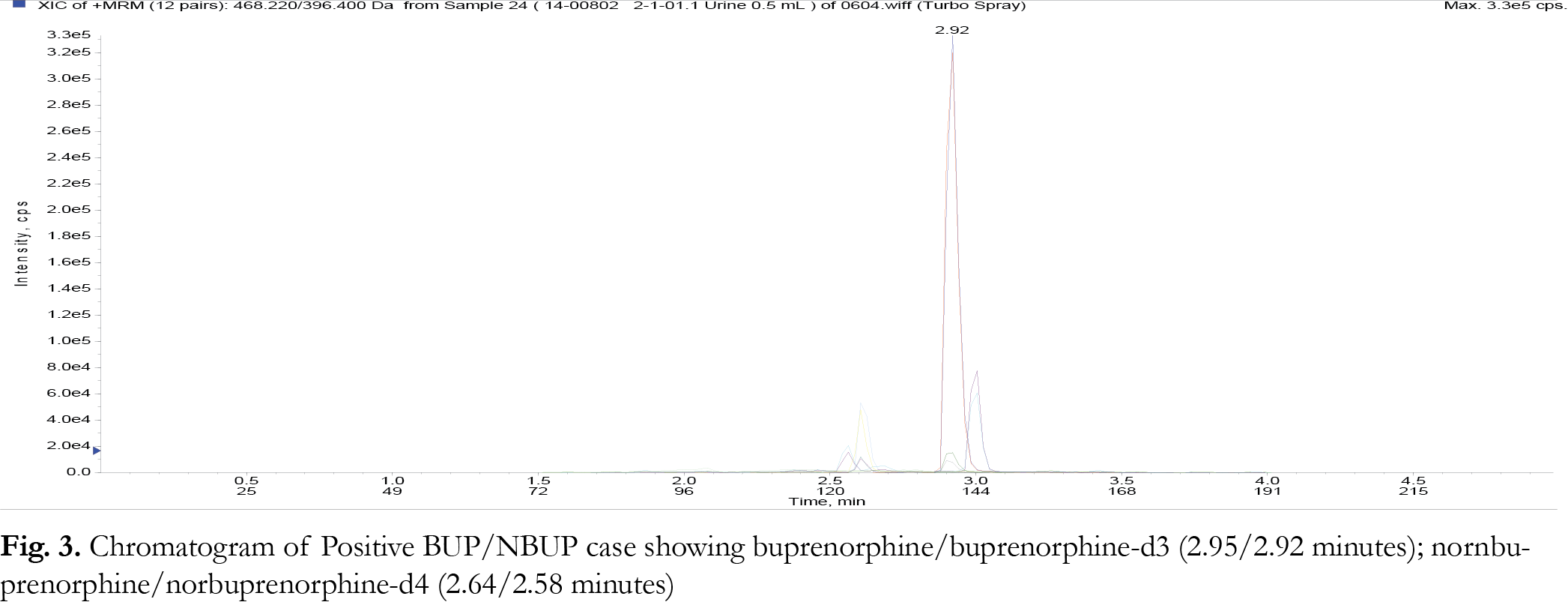
Liquid chromatography was performed in gradient mode employing the Selectra ® DA column and a mobile phase consisting of acetonitrile and 0.1% aqueous formic acid at a flowrate of 0.5 mL/minute. The injection volume was 10 µL for each analytical run. The retention times for the parent compounds were found to be: buprenorphine/ buprenorphine-d3 (2.95/2.92 minutes); nornbuprenorphine/norbuprepnorphine-d4 (2.64/2.58 minutes); naloxone (1.85 minutes). The gradient program is shown in {Tablink Table 1}Table 1.{/Tablink}
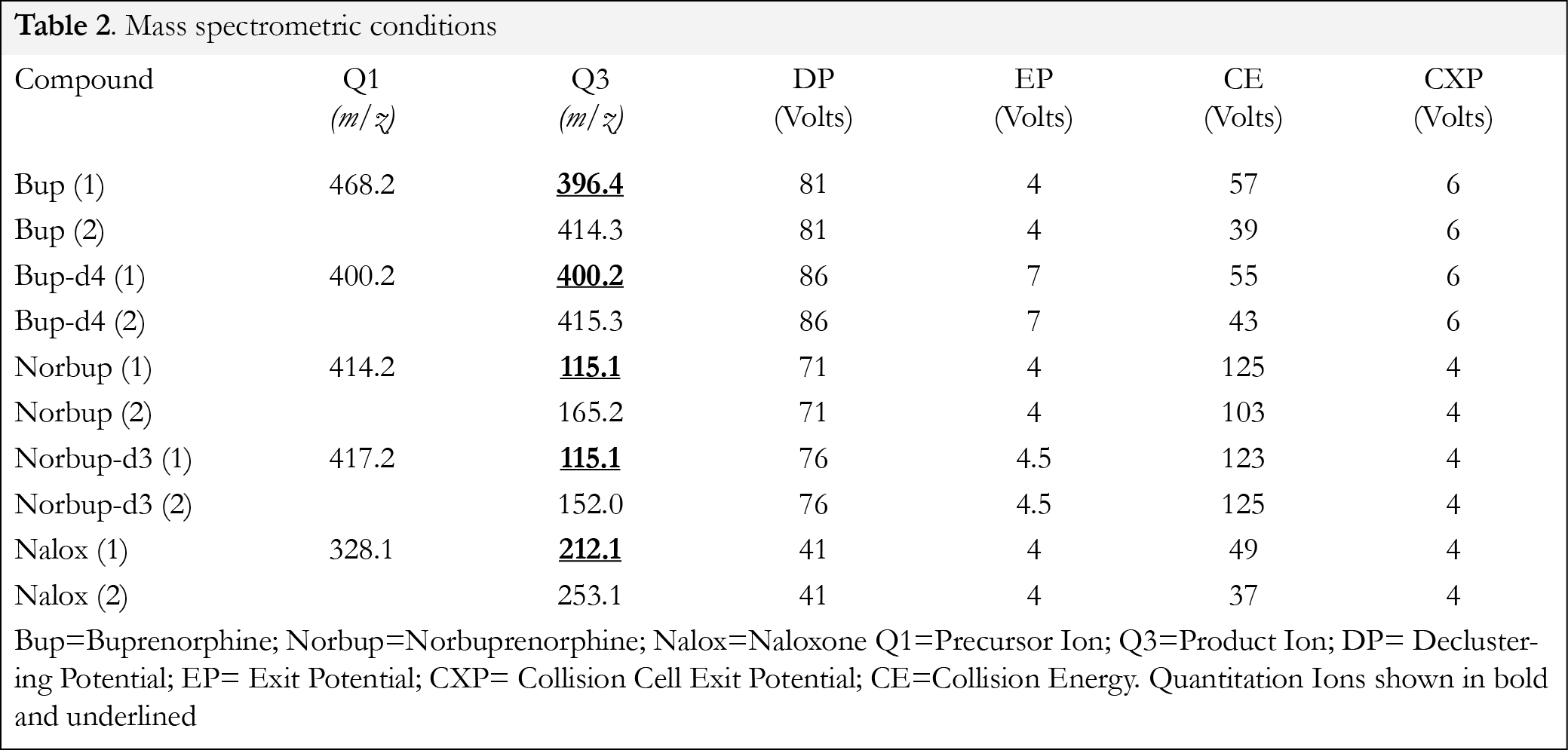
Tandem mass spectrometric analysis was performed in positive multiple reaction monitoring (MRM) mode for:/buprenorphine-d4, norbuprenorphine/norbuprenorphine-d3, and naloxone. The pentodeuterated internal standard for naloxone was not available to the authors at time of study. The mass spectrometric conditions are shown in Table 2. Tandem mass spectrometry was performed under the following conditions: curtain gas setting= 15, collision gas setting= medium, ion spray voltage setting= 5000V, temperature setting= 650°C, ion source gas #1 setting= 50, ion source gas #2 setting= 50. The analytical data was collected using Analyst Software Version 1.5.2 supplied by Applied Biosystems (Now ABSCIEX). Tandem mass spectral conditions including quantitation ions are presented in Table 2.
Positive confirmation of the analytes was based upon peak retention time and the ratios of the MRM. Retention time had to be within 0.2 minutes of the lowest standard, and ratio of the two transitions had to be less than 25%.
Sample Preparation for analysis
Calibrators and controls
Calibrators were prepared by the addition of 10, 50, 100, 250, 500, and 1000 ng of BUP, NBUP and NALOX into 1 mL samples of drug free urine samples. To these samples 500 ng of the internal standard solution (BUP-d3, NBUP-d4) was added. This involved adding 50µL of a solution containing 10µg/ mL of BUP-d3/NBUP-d4) to each sample. Control samples were prepared by the addition of 40 ng and 800 ng of BUP, NBUP and NALOX to 1 mL of drug free urine samples in addition to 500 ng of the internal standard solution. Test samples were chosen from BUP positive urine samples that had been previously analyzed and reported for the BUP. These samples were completed, closed and marked for disposal according to current MSPCL procedures. These samples were prepared by adding 500 ng of internal standard solutions to 1 mL aliquots of urine. All determinations were performed in duplicate. A negative control sample was prepared by the addition of only the internal standard (500 ng) to a sample of drug free urine (1 mL).
To each sample (calibrator, control and test sample) was added: 1 mL of pH 5 acetate buffer and 50 µL of Selectrazyme β-glucuronidase and vortex mixed for approximately 30 seconds before being incubated for 1 hour at 65°C in a thermostatically controlled water bath. The samples were then cooled to room temperature. Calibrators, control samples, and test samples were treated in an identical mode with regard to sample extraction i.e. after buffering with 3 mL of phosphate buffer (pH 6, 0.1M), the samples were vortex mixed for approximately 1 minute then centrifuged at 3000 rpm for 10 minutes. The supernatant liquid was applied to a pre-conditioned SPE cartridge. To assess the performance of the procedure, calibration curves were constructed twice daily over five consecutive days using the spiked controls, from this data intra-day and inter- day values were obtained. The data was obtained by using 2 replicates per concentration level for the construction of each calibration curve, and 2 replicates for each of the controls for assessment of precision and accuracy. The control samples had to be within ±20% of their nominal value.
Solid Phase Extraction
Solid phase extraction columns were conditioned by the sequential addition of: 1 x 3 mL of methanol, 1 x 3 mL of DI water, and 1 x 1 mL of 0.1 M phosphate buffer (pH 6). Each liquid was allowed to percolate through the sorbent using gravity without allowing the sorbent to dry out in between steps.
Following the passage of the methanol, DI water and 0.1 M phosphate buffer (pH 6) through the SPE columns, each diluted sample (i.e. calibrator, control, and case item) was loaded on to an individually marked SPE tube, and allowed to pass through the sorbent using gravitational flow. The SPE columns were then washed with: 1 x 3 mL of DI water, 1 x 1 mL of 1.0 M acetic acid, and 1 x 3 mL of methanol, respectively. The SPE columns were then dried for 10 minutes by applying a vacuum to the SPE manifold at 15 inches of mercury pressure via an electric vacuum pump.
The analytes were eluted from the SPE columns by the addition of 1 x 3 mL of a solution consisting of methylene chloride-isopropanol-ammonium hydroxide (78:20:2). This solution was prepared daily by adding 2 mL of concentrated ammonium hydroxide solution to 20 mL of isopropanol and mixing well. To this solution was added 78 mL of methylene chloride, and the resultant solution was transferred to a clean screw top bottle for use. A screw top bottle ensures that the basicity of the solution remains high by eliminating any loss of ammonia from the bottle. The elution solvent was allowed to flow through the SPE sorbent with the aid of gravity and collected in separate glass tubes (75 mm x 12 mm). The eluates were evaporated to dryness under a gentle stream of nitrogen at 35 °C. The residue was dissolved in 100 μL of a solution containing 95% of mobile phase component (MPA) and 5% of mobile phase component (MPB). This solution was transferred to an autosampler vial containing a low volume insert (100 μL) for analysis by LC-MS/MS.
Matrix Effects
Studies into the matrix effects were performed according to a previously published procedure (34). In this part of the study, aliquots of the BUP/NBUP/NALOX solution (covering the linear range) were evaporated to dryness at 35°C using a gentle stream of nitrogen and dissolved in 100 μL of a solution containing 95% of mobile phase solvent MPA and 5% mobile phase solvent MPB. Each of the solutions were evaporated to the mobile phase and analyzed by LC-MS/MS (A). Concurrently, a set of urine samples obtained from five different sources were subjected to the SPE process noted, after elution of the SPE columns, the elution solvent was fortified with BUP/NORBUP/NALOX (covering the linear range) and evaporated to dryness before being dissolved in 100 μL of 95% (MPA) and 5% (MPB). A second set of urine samples similarly obtained from five different sources were fortified with BUP/NORBUP/NALOX (covering the linear range) and processed via the SPE method. After elution and evaporation to dryness, 100 μL of mobile phase solution (MPA and MPB) was added to dissolve the residue (C). The data (peak areas) for A, B, and C were collected by Analyst 1.5.2. By comparing the peak areas of B with those of A an assessment of matrix effects was made. The comparison of peak areas for C with B provided data for the recoveries.
A solution of BUP/NBUP/NALOX (concentration: 50 ng/ mL) was infused into the tandem mass spectrometer using the on board syringe pump (controlled by Analyst 1.5.2 software) via a Hamilton syringe (model# 1001TLL, 1 ml volume) (supplied by Fisher Scientific) at a flowrate of 5 µL/ minute. At the same time as the solution of BUP/NBUP/NALOX was flowing into the mass spectrometer, a 10 µL aliquot of the SPE extracted urine matrix (samples of urine confirmed to contain no drug material) was injected using the autosampler syringe on the Shimadzu liquid chromatograph. The liquid chromatograph and mass spectrometer were arranged so that samples from the liquid chromatograph were mixed into the flow of BUP/NORBUP/NALOX and metabolites via a 3 port T section before the total flow entered the mass spectrometer. Any suppression effects on the BUP/NBUP/NALOX could be monitored at the MRM’s for the noted drugs.
Selectivity
In analyzing samples of urine extracts via SPE and LC-MS/MS it is essential to ensure that the interfering effects of other drug compounds can be eliminated. In this procedure, samples of urine extracts were spiked with a cocktail of drugs at a concentration equivalent to of 100 ng/ mL of urine sample: (bupropion, lidocaine, methadone, amitriptyline, nortriptyline, thioridazine, trazodone, mesoridazine, meperidine, diphenhydramine, phenyltoloxamine, imipramine, desipramine, benztropine, trimethoprim, diltiazem, haloperidol, strychnine, morphine, codeine, 6-acetylmorphine, oxycodone, oxymorphone, hydrocodone, noroxycodone, hydromorphone, diazepam, nordiazepam, oxazepam, temazepam, alprazolam, α-hydroxyalprazolam, lorazepam, triazolam, α-hydroxytriazolam, flunitrazepam, 7-amino-flunitrazepam, chlordiazepoxide, midazolam, α-hydroxymidazolam, flurazepam, desalkyl-flurazepam, clonazepam, 7-amino-clonazepam, cocaine, benzoylecgonine, cocaethylene, ecgonine, ecgonine methyl ester, ecgonine ethyl ester) and extracted according to the SPE method.
Results and discussion
Recovery
It was found that the mean recovery of BUP, NBUP, and NALOX from drug free urine samples was determined to be BUP 92±5%, NBUP 95 ±5%, and NALOX 91 ±5%, respectively. This is an excellent indicator for the efficiency of the extraction procedure of the compounds using urine as a matrix. This procedure was as performed twice daily over a period of five days.
Imprecision of Analysis
The results of the analysis of the spiked control samples of urine: (40 ng/ mL, 800 ng/ mL, respectively) are shown in Table 3. Analysis of the control samples was performed at the same time as the calibration curves were constructed i.e. over a period of five days. Control samples were prepared by adding the BUP/NBUP/NALOX to drug free urine samples (1 mL) and treating as per the test samples.
Intra-day variation for the analysis was found to be less than 8%. The inter-day variation for the analysis was found to be less than 12%. Ion suppression studies revealed that suppression of monitored ions was less than 6 %. This method was found to be linear (r2> 0.995) over the dynamic range 10 ng/ mL to 1000 ng/ mL.
LOD/LOQ
The limit of detection (LOD) of a particular method can be defined as the level at which the signal to noise ratio for the particular analyte is greater than or equal than 3:1. The limit quantification (LOQ) for the method is the level at which the signal to noise ratio for a particular analyte is greater than or equal to 10:1. In this study, LOD values were determined empirically by analyzing extracted samples of drug free urine fortified with BUP/NBUP/NALOX by LC-MS/MS according to the SPE method. This was performed until the lowest level at which each of the respective analytes just failed the signal to noise ratio of 3:1. This was observed to be 0.5 ng/ mg. In terms of LOQ, samples of drug free urine samples were spiked with the noted drugs at concentrations below 10 ng/ mg and extracted according to the SPE procedure until the analytes could just failed a signal to noise ratio of 10:1; this value was found to be 1.0 ng/ mL. Representative chromatograms at LOQ and genuine urine samples are shown in Figures 1-3 . None of the analyzed samples were found to contain BUP or NBUP at concentrations less than 10 ng/ mL, thus the lowest calibrator was set to 10 ng/ mL not 1 ng/ mL.
Selectivity
It was observed that the interfering effects of the cocktail of spiked drug compounds was not found to be significant.
Solid Phase Extraction
In this procedure, dilution of the sample of drug free urine with 3 mL of an aqueous pH 6 buffer permits both efficient flow and optimal sorbing of the drugs onto the SPE sorbent. In employing a mixed mode (C8 and strong cation exchange chemistries), the sample can be cleaned up by washing the sorbent with DI water, aqueous acetic acid and methanol which leaves the drugs in a much cleaner state than when they were originally applied to the SPE column. This effect is noted in the low matrix effects and ion suppression values. This procedure permits the analytes to be efficiently eluted from urine samples using a relatively mid polar basic solvent system that is easily evaporated to dryness at less than 40°C.



Tandem Mass Spectrometry
In this methodology, LC-MS/MS has been successfully applied to the extraction and analysis of buprenorphine and its metabolite as well as naloxone rather than GC-MS where a derivatization procedure (i.e. reaction with a silyl reagent (e.g. BSTFA) or an acyl derivative [22] required not only to quantify, but also confirm the identity of the compounds as the authors have found in their own laboratories that BUP, NBUP, and NALOX compounds do not chromatograph well if at all on GC-MS in their underivatized state. By employing LC –MS/MS with specific MRM’s, the individual drugs can be targeted, confirmed, and quantified in urine samples without this use of derivatization. This efficient MS/MS procedure coupled with a short LC method offers analysts the ability of determine concentrations of the noted drugs within a short turnaround time. The hydrolysis procedure is a required step in order to cleave the glucuronide moiety from the individual compounds and present the parent compounds for detection/quantification by tandem mass spectrometry.
Case samples
This method offers the toxicological analyst in a forensic setting the ability to provide information regarding the use of naloxone in buprenorphine cases thus differentiating between these and Suboxone® ones. Data for genuine cases presenting concentrations for the BUP/NBUP/NALOX in urine are shown in Table 4. In case samples # 4, 6, 7, and 10 no naloxone was observed, while in cases 1-3,5,8-9 naloxone was observed indicating the possibility that a BUP/NALOX medication was administered. The data presented in Table 3 also indicates the total concentrations of BUP/NBUP/NALOX in the urine, the degree of metabolism cannot be estimated without separation of the parent from the glucuronide. In all these cases the concentrations of BUP was greater than NBUP indicating that the metabolite of BUP exists as the predominant species in urine. These cases were previously analyzed for BUP/NBUP but not for NALOX. Since the cases presented here involve the analysis of ante-mortem urine samples taken from drivers suspected of operating a motor vehicle under the influence of drugs, the naloxone could not administered by emergency personnel as the driver is in the custody of a law enforcement agent upto, during and after the urine sampling. Any contemporaneous medical intervention would be noted in the records.
Conclusion
This study shows that enzymatic hydrolysis, solid phase extraction, and LC-MS/MS can be employed efficiently to analyze BUP, NBUP, and NALOX at the same time in urine samples. The use of SPE provides analysts with clean extracts that can be separated and quantified by LC-MS/MS rapidly, thus providing an efficient procedure for use by forensic toxicological analysts involved in drugs and driving cases. The use of tandem mass spectrometry permits targeted analysis which allows toxicologists to differentiate between cases involving buprenorphine administration and those cases in which buprenorphine and naloxone have both been used.
References
1. Blum K, Oscar-Berman M, Femino J et al. Withdrawal from buprenorphine/naloxone and maintenance with a natural dopaminergic agonist: a cautionary note. J Addict Res Ther 4 (2), 1-25, (2013). [CrossRef]
2. Zubieta J, Greenwald MK, Lombardi U et al. Buprenorphine-induced changes in mu-opioid receptor availability in male heroin-dependent volunteers: a preliminary study. Neuropsychopharmacology 23, 326-334 (2000). [CrossRef]
3. Campbell ND, Lovell AM, The history of the development of buprenorphine as an addiction therapeutic. Ann NY Acad Sci1248, 124-139 (2012). [CrossRef]
4. Cone EJ, Godordetzky CW, Yousefnejad D et al., The metabolism and excretion of buprenorphine in humans. Drug Metab Disp 12 (5), 577-581 (1984).
5. Chang Y, Moody DE, McCance-Katz EF, Novel metabolites of buprenorphine detected in human Liver Microsomes and human urine. Drug Metab Disp 34(3), 440-448 (2006). [CrossRef]
6. Ohtani M, Kotaki H, Sawada Y et al. Comparative analysis of buprenorphine- and norbuprenorphine-induced analgesic effects based on pharmacokinetic-pharmacodynamic modeling. J Pharmacol Exp Ther 272, 505-510 (1995).
7. Ohtani M, Kotaki H, Nishitateno K et al. Kinetics of respiratory depression in rats induced nu buprenorphine and its metabolite norbuprenorphine. J Pharmacol Exp Ther 281, 428-433 (1997).
8. Gopal S, Tzeng T-Z, Cowan A, Characterization of the pharmacokinetics of buprenorphine and norbuprenorphine in rats after intravenous bolus administration of buprenorphine. Eur J Pharm Sci 15, 287-293 (2003). [CrossRef]
9. Woodhall KL, Chow, BLC, Lauwers A et al. Toxicological findings in fatal motor vehicle collisions in Ontario, Canada: A one year study. J Forensic Sci 60, 669-674 (2015). [CrossRef]
10. Baselt RC, in Disposition of Toxic Drugs And Chemicals in Man 9th Edition, Biomedical Publications, Seal Beach, California, USA, pages 212-214 (2011).
11. Buprenorphine package insert product #336UR, Immunalysis Corp, Pomona, CA, USA (2014).
12. Buprenorphine package insert product MTA-12TM, Venture Labs, Redwood City, CA USA (2014).
13. Pergolizza J, Aloisi AM, Dahan A et al. Current knowledge of buprenorphine and its unique pharmacological profile. Pain Practice 10, 428-450 (2010). [CrossRef]
14. Ishiyama D, Jones J, Towards evidence based emergency medicine: best BETs from the Manchester Royal Infirmary. BET 3: Is nebulised naloxone effective in opioid overdose? EMJ 30 (10) 860 (2013). [CrossRef]
15. Lillsunde P, Korte Comprehensive drug screening in urine using solid-phase extraction and combined TLC and GC/MS identification. J Anal Toxicol 15, 78-81 (1991). [CrossRef]
16. Fuller DC, A simple gas chromatography–mass spectrometryprocedure for the simultaneous determination of buprenorphine and norbuprenorphine in human urine. J Anal Toxicol 32, 626-630 (2008). [CrossRef]
17. El-Sohly, M, Gul W, Feng S et al. Hydrolysis of conjugated metabolites of buprenorphine II. The quantitative enzymatic hydrolysis of norbuprenorphine-3-p-D-glucuronide in Human Urine. J Anal Toxicol 29, 570-573 (2005). [CrossRef]
18. Bottcher M, Beck O, Evaluation of Buprenorphine cedia assay versus GC-MS and ELISA using urine Samples from Patients in substitution treatment. J Anal Toxicol 29, 769-776, (2005). [CrossRef]
19. Alves M, Piccinotti A, Tameni S et al. Evaluation of buprenorphine LUCIO immunoassay versus GC–MS using Urines from a workplace drug testing program. J Anal Toxicol 37,175–178 (2013). [CrossRef]
20. George S, George C, Chauhan M, The development and application of a rapid gas chromatography–mass spectrometry method to monitor buprenorphine withdrawal protocols. Forensic. Sci Int’l143, 121-125 (2004). [CrossRef]
21. Wang G, Vincent M, Rodigues W, Development and GC-MS Validation of a highly sensitive recombinant G6PDH-based homogeneous immunoassay for the Detection of buprenorphine and norbuprenorphine in urine. J Anal Toxicol 31, 377-382 (2007). [CrossRef]
22. Wu C-H, Yang S-C, Wang Y-S et al. Evaluation of various derivatization approaches for gas chromatography–mass spectrometry analysis of buprenorphine and norbuprenorphine. J Chromatogr A 1182, 93-112 (2008). [CrossRef]
23. Concheiro-Guisan M, Shakelaya DM, Huestis MA et al. Simultaneous quantification of buprenorphine,norbuprenorphine, buprenorphine glucuronide, andnorbuprenorphine glucuronide in human placenta by liquid chromatography mass spectrometry. Anal Bioanal Chem. 394 (2), 513-522 (2009). [CrossRef]
24. Kacinko SL, Concheiro-Guisan M, Shakelaya DM et al. Development and validation of a liquid chromatography–tandem mass spectrometry assay for the simultaneous quantification of buprenorphine, norbuprenorphine, and metabolites in human urine. Anal Bioanal Chem 392(5), 903-911 (2008). [CrossRef]
25. Kacinko SL, Jones HE, Johnson RE et al. Urinary excretion of buprenorphine, norbuprenorphine, buprenorphine-glucuronide, and norbuprenorphine-glucuronide in pregnant women receiving buprenorphine maintenance treatment. Clin Chem 55(6), 1177-1187 (2009). [CrossRef]
26. McMillan GA, Davis R, Carlisle H et al. Patterns of free (Unconjugated) buprenorphine, norbuprenorphine, and their glucuronides in urine using liquid chromatography–tandem mass Spectrometry. J Anal Toxicol 36, 81-87 (2012). [CrossRef]
27. Kronstrand R, Nystrom I, Andersson M et al. Urinary detection times and metabolite/parent compound ratios after a single dose of buprenorphine. J Anal Toxicol 32, 586-593 (2008). [CrossRef]
28. Al-Asmari AI, Anderson R, Comparison of nonhydrolysis and hydrolysis methods for the determination of buprenorphine metabolites in urine by liquid chromatography tandem mass spectrometry. J Anal Toxicol 32, 744-753 (2008). [CrossRef]
29. Harris DS, Jones RT, Welm S et al. Buprenorphine and naloxone co-administration in opiate-dependent patients stabilized on sublingual buprenorphine. Drug Alcohol Dep 61, 85-94 (2000). [CrossRef]
30. Nasser AF, Heidbreider C, Liu Y et al. Pharmacokinetics of sublingual buprenorphine and naloxone in subjects with mild to severe hepatic impairment (Child-Pugh Classes A, B, and C), in Hepatitis C Virus-Seropositive subjects,and in healthy Volunteers. Clin Pharmacokinet. DOI 10.1007/s40262-015-0238-6.
31. Heikman P, Hakkinen M, Gergov M et al. Urine naloxone concentration at differentphases of buprenorphine maintenance treatment. Drug Test Anal 6, 220-225 (2014). [CrossRef]
32. Tzatzarakis MN, Vakonaki E, Kovatsi L et al. Determination of buprenorphine, norbuprenorphine and naloxone in fingernail clippings and urine of patients under Opioid substitution therapy. J Anal Toxicol 39, 313-320 (2015). [CrossRef]
33. Harris DS, Mendelson JE, Lin ET et al. Pharmacokinetics and subjective effects of sublingual buprenorphine, alone or in combination with naloxone lack of dose proportionality. Clin Pharmacokinet 43(5), 329-340 (2004). [CrossRef]
34. Matuszewski BK, Constanzer M, Chavez-Eng CM, Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC−MS/MS. Anal Chem 75, 3019-3030 (2003). [CrossRef]
All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License.
![]()
 Open Access Article
Open Access Article Peer-reviewed Article
Peer-reviewed Article Creative Commons Attribution 4.0 License
Creative Commons Attribution 4.0 License